Our current paradigm of insulin resistance is that of a lock and key, and it’s simply wrong.
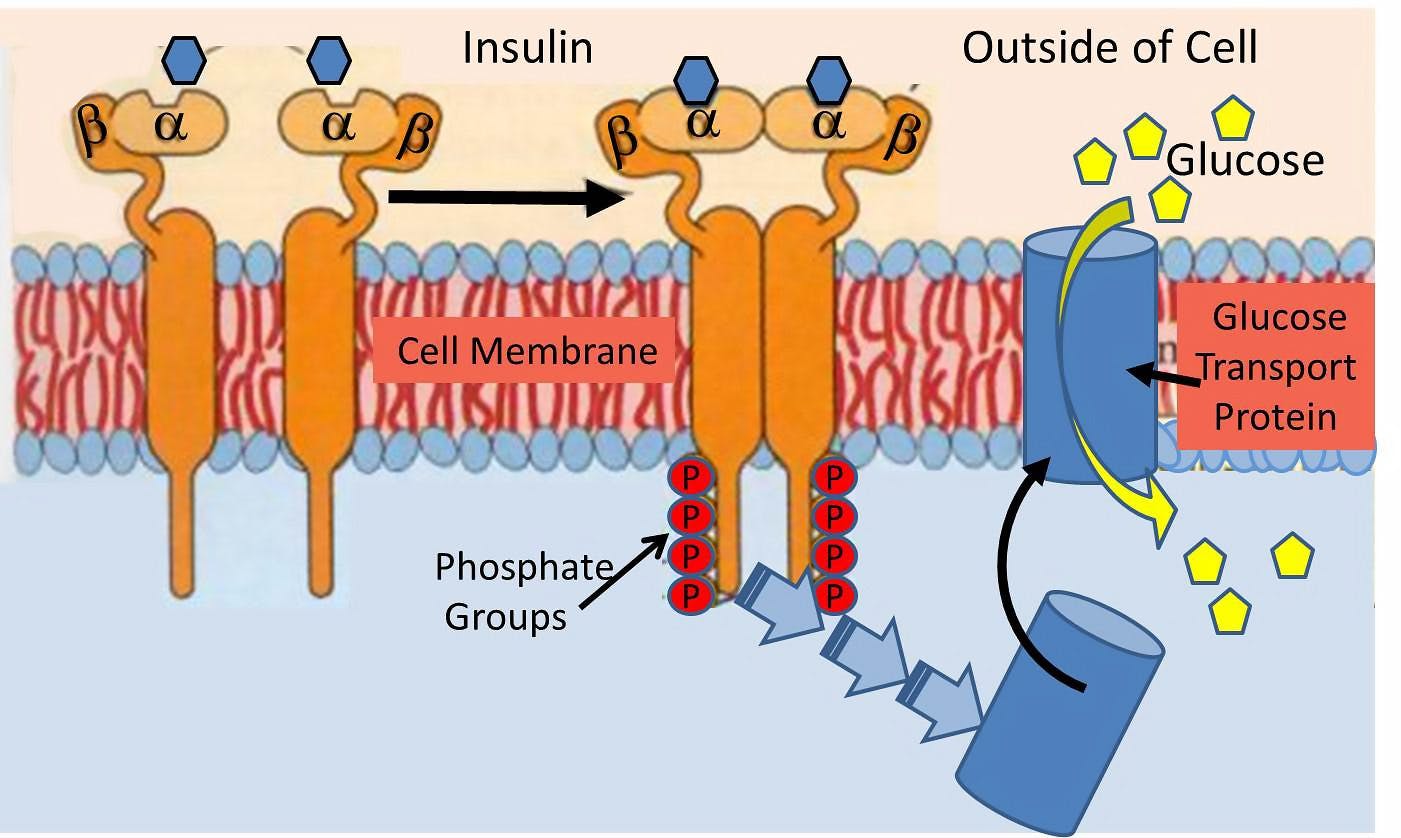
Insulin is a hormone that acts upon a hormonal receptor on a cell surface in order to have an effect. This is often referred to as lock and key model.
The lock is the insulin receptor which keeps the gates to the cell closed. When the proper key (insulin) is inserted, then the gate opens to let glucose from the blood inside the cell. This glucose is then able to power the cell machinery.
Once you remove the key (insulin) then the gate closes back up and glucose in the blood is no longer able to go inside the cell.
Lock and key during insulin resistance
What happens during the phenomenon of insulin resistance? Classically, we imagine that the lock and key no longer fit very well. The key (insulin) is able to open the lock (receptor) but only partially and not very well. As a result, the glucose is not able to pass through the gate normally.
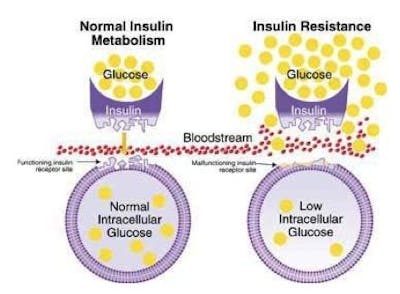 This results in lower than normal amounts of glucose inside the cell. The glucose, which is now blocked by the closed gate, piles up outside the cell in the blood, which we can detect as elevated blood sugar and make the clinical diagnosis of type 2 diabetes.
This results in lower than normal amounts of glucose inside the cell. The glucose, which is now blocked by the closed gate, piles up outside the cell in the blood, which we can detect as elevated blood sugar and make the clinical diagnosis of type 2 diabetes.
This has also been described as a state of internal starvation since the cell has little glucose on the inside. The knee-jerk reaction is for the body to increase production of insulin (key). Since each key works less well than previously, the body over-produces the number of keys to make sure that enough glucose goes into the cells. A nice neat theory.
The problems
The problem, really, is that this paradigm does not really fit reality. First, is the problem the insulin, or the insulin receptor? Well, it’s really quite easy these days to look at the structure of insulin and the structure of the insulin receptor of insulin resistance patients. You simply isolate the insulin or some cells and check their structure with fancy molecular tools. It immediately becomes clear that there is nothing wrong with either the insulin or the receptor. So what’s the deal?
The only remaining possibility is that there is something that is gumming up the system. Some kind of blocker that interferes with mechanism of the lock and key. But what? There’s all kinds of theories. Inflammation. Oxidative Stress. Advance glycation End Products. All the usual buzzwords that come out when doctors have really no idea. With this model, we have no real friggin’ idea what caused the insulin resistance. Without understanding what causes IR, we have no chance of treating it.
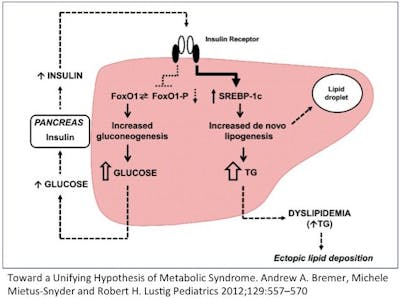 Then there’s the central paradox of hepatic insulin resistance. Let me explain. Insulin has two major actions on the liver. Remember that insulin goes up when you eat. It tells the body to stop producing glucose in the liver (gluconeogenesis) because there is lots of glucose coming in from the stomach (food). This is mediated through the FOX01 pathway.
Then there’s the central paradox of hepatic insulin resistance. Let me explain. Insulin has two major actions on the liver. Remember that insulin goes up when you eat. It tells the body to stop producing glucose in the liver (gluconeogenesis) because there is lots of glucose coming in from the stomach (food). This is mediated through the FOX01 pathway.
The second major action in the liver is to increase the production of fat (De Novo Lipogenesis (DNL)). This is to deal with the incoming flood of glucose that the body can’t use right way. This is mediated through the SREBP-1c pathway.
So, if the liver becomes insulin resistant, then the effect of insulin should drop for both of these actions. That is, the liver should continue to make glucose, and stop making fat. But that’s only the case for gluconeogenesis. That is, during insulin resistance, the liver continues to make new glucose as expected. But DNL (making new fat) continues and actually increases. So insulin’s effect on DNL is not blunted but accelerated!
What the hell?
How in seven hells can this insulin resistant liver selectively be resistant to one effect of insulin yet accelerate the effect of the other? In the very same cell, in response to the very same levels of insulin, with the very same insulin receptor? That seems crazy. The same cell is insulin resistance and insulin sensitive at the same time!
A better explanation: overflow
How can we explain this paradox?
We need a new paradigm of insulin resistance that better fits the facts. In fact, we can think of insulin resistance as an overflow phenomenon, instead of a lock and key one. All we really know about insulin resistance is that it is much more difficult to move glucose into an ‘insulin resistant’ cell than a normal one.
But this does not necessarily mean that the door is jammed. Instead, perhaps the cell is already overflowing with glucose and therefore more glucose cannot go in.
Imagine the cell to be a subway car. When the door opens, the passengers on the outside (glucose in the blood) march in a nice orderly manner into the empty subway car (cell). Normally, it doesn’t really require much of a push to get this glucose into the cell (insulin gives the push).
But during insulin resistance, the problem is not that the door does not open. The problem, instead is that the subway car (cell) is already overflowing with passengers (glucose). Now the glucose outside the cell simply can’t get in and is left crowded on the platform.
 Insulin tries to push the glucose into the cell like the Japanese Subway Pushers, but they simply can’t do it because it’s full. So, it looks like the cell is resistant to the effects of the insulin, but really the problem is that the cell is already overflowing. So, the knee jerk reaction is to manufacture more insulin (pushers) to help push glucose into the cell. Which works, but only for a while.
Insulin tries to push the glucose into the cell like the Japanese Subway Pushers, but they simply can’t do it because it’s full. So, it looks like the cell is resistant to the effects of the insulin, but really the problem is that the cell is already overflowing. So, the knee jerk reaction is to manufacture more insulin (pushers) to help push glucose into the cell. Which works, but only for a while.
So, the cell is not in a state of ‘internal starvation’. Instead, the cell is overflowing with glucose. Glucose starts spilling out into the cell, which looks like gluconeogenesis has not been stopped consistent with insulin resistance. But what happens to fat production?
In the classic model of insulin resistance, the paradox was that DNL was enhanced, not decreased which looked a lot like heightened insulin sensitivity instead of resistance. But in the overflow model, the DNL would be enhanced because the cell is trying to rid itself of the excess glucose by producing extra fat. The cell is overflowing and not in an ‘internal starvation’ mode.
Why it matters
Why is this critically important? Because understanding this new paradigm will lead to the answer of how insulin resistance develops and what we can do about it. The problem does not lie with either insulin nor the insulin receptor. Both are normal. The problem is that the cell is completely stuffed full of glucose. So, what caused it?
The answer then seems obvious – it’s a matter of too much glucose and too much insulin. In other words, it was the insulin itself that caused the insulin resistance. We don’t need to chase shadows looking for some mysterious cause of insulin resistance.
Once we understand that excessive glucose and excessive insulin is the cause of the insulin resistance, then we can now devise a rational treatment. Reduce insulin and reduce glucose. Once you reverse the insulin resistance, you cure the type 2 diabetes.
A better way
How to Reverse Type 2 Diabetes
Earlier by Dr. Fung
Why the First Law of Thermodynamics Is Utterly Irrelevant
How to Fix Your Broken Metabolism by Doing the Exact Opposite
The Biggest Loser FAIL and That Ketogenic Study Success
Videos
 4.6 out of 5 stars5 star78%4 star9%3 star5%2 star3%1 star1%104 ratings10445:20
4.6 out of 5 stars5 star78%4 star9%3 star5%2 star3%1 star1%104 ratings10445:20 4.8 out of 5 stars5 star90%4 star4%3 star0%2 star2%1 star2%41 ratings4138:45
4.8 out of 5 stars5 star90%4 star4%3 star0%2 star2%1 star2%41 ratings4138:45
More with Dr. Fung
Dr. Fung has his own blog at intensivedietarymanagement.com. He is also active on Twitter.
His book The Obesity Code is available on Amazon.

The post A New Paradigm of Insulin Resistance appeared first on Diet Doctor.
http://www.dietdoctor.com/a-new-paradigm-of-insulin-resistance
No comments:
Post a Comment